fenyloketonuria - klasyczne objawy transmitowanego dziedziczne i diety
Spis treści
- 1Jak się okazuje, fenyloketonuria choroby ,
- 2Mechanizm choroby ,
- 3 , fenyloketonuria u dzieci ,
- 4Objawy ,
- 5 , czynniki strącające przyczyny i ,
- 6Diagnostyka
- 7Leczenie klasycznych fenyloketonurię ,
- 8Cechy i noworodków odżywianie diety ,
- 9Dieta dla dzieci w wieku przedszkolnym i szkolnym dzieci
- 10Grupy wyrobów z PKU ,
- 11Sposób kontroli poziomu fenyloalaniny we krwi ,
- 12Filmy wideo
Wystąpienie choroby wiąże się z wadami genetycznych urządzenia komórkowego - fenyloketonuria - znajduje się lista kilku chorób dziedzicznych do leczenia. Pionierem choroby był lekarz Norwegia, IA Fellynh później okazało się, że rozwój choroby, a jedynym genem odpowiedzialnym zwany fenilalaninhidroksylazy genomu (chromosom długie ramię 12oy zawierające do 4,5% całkowitej materiału DNA komórki). Dziedziczne wady w wyniku częściowej lub całkowitej inaktywacji enzymu wątrobowego fenyloalaniny-4-hydroksylazy.
Jeżeli okaże się, że fenyloketonuria choroby ,
choroby dziedziczne fenyloketonuria (PKU) prowadzi do przewlekłych zatrucia organizmu substancji toksycznych utworzonych wskutek upośledzenia metabolizmu aminokwasów i procesówhydroksylacja fenyloalaniny. Stała zatrucie powoduje uszkodzenie ośrodkowego układu nerwowego (OUN), która działa jako przejaw postępującego spadku Intelligence (fenylpyrovynohradnaya upośledzeniem umysłowym).
Choroba ścięgna przejawia się nadmiernym gromadzeniem się fenyloalaniny w organizmie i produktami jej niewłaściwego metabolizmu. Do innych czynników dotyczy fenyloketonuria podniesione transportu aminokwasów przez BBB, niewielkiej liczbie (neuroprzekaźników serotoniny, histaminy, dopaminy). W przypadku braku szybkiego leczenia choroby prowadzi do upośledzenia umysłowego i może spowodować śmierć dziecka.

Mechanizm rozwoju choroby
czynników Prychynoobrazuyuschym genetycznej zaburzeń metabolicznych stanowi blok, który zapobiega tworzeniu się fenyloalaniny (4-hydroksylazę enzymu odpowiedzialnego za konwersję aminokwasu tyrozyny w fenilaninu). Proteynohennaya aminokwasu tyrozyny jest składnikiem białka i pigmentu melaniny, co jest niezbędne do funkcjonowania wszystkich systemów organizmu, a jego brak prowadzi do fermentopathy.
W wyniku hamowania tworzenia metabolitu indukowanego mutacyjnej dezaktywacji aktywacji enzymu wspomaga wymianę fenyloalaniny. Aromatyczny alfa-aminokwas, w wyniku wadliwych procesów wymiany, rozpada się na toksyczne pochodne, które nie powstają w normalnych warunkach:
- kwas fenylopirogronowy (fenylo pirogronian) - tłuszczowy aromatyczny alfa-ketokwas, jego wykształcenieprowadzi do mielinizacji procesów neuronalnych i demencji;
- kwas fenylomorficzny - produkt powstały podczas regeneracji kwasu fenylo-wirionowego;
- Fenyloetyloamina - początkowy związek dla biologicznie aktywnych przekaźników impulsów elektrochemicznych, zwiększa stężenie dopaminy, adrenaliny i norepinefryny;
- Ortofenylooctan - substancja toksyczna, która powoduje naruszenie procesów metabolicznych związków kałowych w mózgu.
Statystyki medyczne pokazują, że patologicznie zmieniony gen występuje u 2% populacji, ale się nie objawia. Defekt genetyczny jest przekazywany z rodzica dziecka tylko na obecność choroby obu partnerów przy dziecku w 50% przypadków staje nosicielem zmutowanego genu, podczas gdy pozostały zdrowe. Prawdopodobieństwo, że fenyloketonuria u noworodków doprowadzi do choroby wynosi 25%.
Według jakiego rodzaju dziedziczenia
Choroba ścięgna jest genetyczną dewiacją, odciśniętą na autosomalnym recesywnym typie. To dziedziczenie oznacza, że rozwój objawów choroby wrodzonej nastąpi tylko wtedy, gdy dziedziczenie wadliwego kopie Geno dziecko od obojga rodziców są heterozygotycznych nosicieli genu zmodyfikowanego.
Rozwój choroby wrodzone w 99% przypadków, mutacja genu odpowiedzialnego za kodujący enzym, który zapewnia syntezę fenyloalanino-4-hydroksylazy (klasyczny fenyloketonuria). Do 1% chorób genetycznych wiąże się ze zmianami mutacyjnymi zachodzącymi w innych powodowanych genachniewystarczalność reduktazy dihydropterydyny (PKU typu II) lub tetrahydrobiopteryny (PKU typu III).
Fenyloketonuria u dzieci
Klasyczna postać choroby narządów płciowych u dzieci w większości przypadków przejawia się na pozornie zauważalnych objawach, od 3-9 miesięcy życia. Noworodki z wadliwym genem wyglądają zdrowo, cechą charakterystyczną jest specyficzny nawyk (wygląd) dziecka. Objaw pojawia się 6-12 miesięcy po urodzeniu.
PKU typu II charakteryzuje się tym, że pierwsze objawy kliniczne pojawiają się po 1,5 roku od momentu wystąpienia światła. Objawy choroby nie ustępują po rozpoznaniu nieprawidłowości genetycznych i rozpoczęciu terapii dietetycznej. Ten rodzaj choroby wrodzonej często prowadzi do śmiertelnego wyniku 2-3 lat życia dziecka. Najczęstsze objawy PKU typu II to:
- wyrażali odchylenia w rozwoju umysłowym;
- hiperrefleksja;
- naruszenie funkcji motorycznych wszystkich kończyn;
- zespół niekontrolowanych skurczy mięśni.
Kliniczne oznaki zmian mutacyjnych w genach typu III są podobne do chorób występujących w typie II. Niedobór tetrahydrobiopteryny charakteryzuje się triadą specyficznych objawów:
- wysoki stopień upośledzenia umysłowego;
- widoczny zmniejszony rozmiar czaszki w stosunku do innych części ciała;
- spastyczność mięśni (z możliwością całkowitej utraty ruchomości kończyn).

Objawy choroby ścięgien
W trakcie badań klinicznych i obserwacji sugerowano, że efekt byłtoksyczny wymiany pochodne fenylalanynovoho powoduje spadek zdolności umysłowych, który jest progresywny i mogą prowadzić do demencji (niedorozwój umysłowy, idiotyzm). Wśród możliwych przyczyn zaburzenia nieodwracalne aktywności mózgu, uważany za odpowiedni ze względu na niższy poziom braku tyrozynowej neuroprzekaźniki, które przesyłają sygnały pomiędzy neuronami.
Dokładna związek przyczynowy pomiędzy chorobą dziedziczną zaburzeń mózgu i do tej pory zidentyfikowano jako mechanizm fenyloketonurię ze względu na stany psychiczne, takie jak эhopraksyya, echolalia, napady złości i drażliwość. Wyniki badań pokazują, że fenyloalanina wywiera bezpośredni toksyczny wpływ na mózg, co może również powodować obniżenie inteligencji.
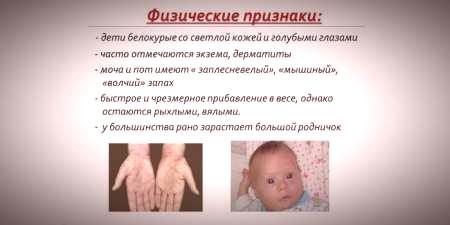
Specyfika struktury i fenotypu
Biorąc pod uwagę, że nasycenie skóry i włosów barwnik w zależności od poziomu tyrozyny w mitochondriach hepatocytów i powoduje fenyloketonuria konwersji zatrzymania fenyloalaniny, pacjenci z tą chorobą mają właściwości fenotypowe (recesywny). Wzmożone napięcie mięśni jest przyczyną zaburzeń w strukturze ciała - to dysplastycheskoe. Do doskonałych cech zewnętrznych fenyloketonurii należą:
- hipopigmentacja - jasna skóra, bladoniebieskie oczy, przebarwione włosy;
- cynizm kończyn;
- zmniejszony rozmiar głowy;
- specyficzną postawę - gdy próbuje stać lub siedzieć dziecko przyjmuje pozę „krawiec” (ręce i nogi zgięte w stawach).
Objawychoroba
W odpowiednim czasie choroba Fellinga ulega skutecznemu leczeniu poprzez dostosowanie żywienia, a rozwój dziecka następuje zgodnie z jego grupą wiekową. Trudność w wykryciu mutacji genu polega na tym, że wczesne objawy są trudne do wykrycia nawet u doświadczonego pediatry. Nasilenie objawów choroby wrodzonej wzrasta wraz z wiekiem dziecka, ponieważ stosowanie żywności białkowej przyczynia się do rozwoju zaburzeń OUN.
Objawy noworodków
W pierwszych dniach życia dziecka objawy zaburzeń patologicznych są trudne do wykrycia - dziecko zachowuje się naturalnie, nie ma opóźnienia w rozwoju. Objawy choroby pojawiają się po raz pierwszy w ciągu 2-6 miesięcy po urodzeniu. Rodzice powinni być czujni na zachowanie dziecka, które charakteryzuje się niską aktywnością, letargiem lub, przeciwnie, lękiem, hiperbolaturą.
Wraz z początkiem karmienia piersią, nowe białka zaczynają dostawać się do ciała noworodka z mlekiem, które służy jako katalizator pojawiania się pierwszych objawów, co wyraźnie wskazuje, że choroba zaczęła się rozwijać. Specyficzne objawy kliniczne choroby obejmują:
- stałe wymioty (często brane za wrodzone zwężenie bramki);
- częste przemieszczanie;
- brak reakcji na bodźce zewnętrzne;
- dystonia mięśniowa (zmniejszone napięcie mięśniowe);
- zespół konwulsyjny (drgawki typu epileptycznego lub nieepileptycznego).
Objawy u dzieci po 6 miesiącach
Jeśli przejaw choroby genetycznej nie jestwystąpił (lub nie był widziany) w ciągu pierwszych 6 miesięcy od narodzin dziecka, a następnie po tym okresie można już dokładnie określić opóźnienie rozwoju psychoruchowego. Objawy zaburzeń genetycznych spowodowanych niedoborem enzymu u dzieci w wieku powyżej sześciu miesięcy to:
- spadek aktywności (do całkowitej obojętności);
- brak prób wstania, siedzenie;
- specjalny "mysi" zapach skóry (zapach pleśni powstaje w wyniku wycofania toksycznych pochodnych fenyloalaniny przez gruczoły potowe i mocz);
- utrata umiejętności wizualizowania twarzy rodziców;
- obieranie skóry;
- pojawienie się zapalenia skóry, egzemy, twardziny skóry.
Postęp choroby w przypadku braku leczenia w dzieciństwie
Jeśli w dzieciństwie nie wykryto odchyleń rozwojowych i nie przeprowadzono odpowiedniego leczenia, choroba zaczyna się aktywnie rozwijać i często prowadzi do niepełnosprawności. Brak leczenia we wczesnym stadium choroby powoduje pojawienie się takich objawów choroby w wieku 1,5 roku:
- małogłowie (zmniejszona wielkość mózgu);
- spływ (przesunięcie górnego rzędu zębów do przodu);
- późniejsza erupcja zębów;
- niedorozwój szkliwa (cienkość lub całkowity brak szkliwa zębów);
- opóźnienie rozwoju językowego aż do całkowitego braku mowy;
- 3, 4 stopnie oligofrenii (upośledzenie umysłowe, upośledzenie umysłowe);
- wrodzona choroba serca (wady struktury mięśnia sercowego, serca,duże statki);
- zaburzenia układu wegetatywnego (akrocyjanoza, zwiększona potliwość, niedociśnienie tętnicze);
- zaparcie.
Przyczyny i czynniki prowokujące
Aby zamanifestować mutację z autosomalnym recesywnym dziedziczeniem, wadliwy gen musi być dziedziczony od obojga rodziców. Choroby genetyczne tego typu występują u tej samej częstotliwości u nowonarodzonych chłopców i dziewcząt. Patogeneza PKU jest spowodowana naruszeniem wymiany fenyloalaniny, która może występować w 3 formach. Leczenie dietoterapią podlega klasycznej fenyloketonurii typu I.
Nietypowe formy choroby można wyleczyć poprzez dostosowanie żywienia. Odchylenia te wynikają z niedoboru tetrahydropteryny, reduktazy dehydropteryny (rzadko - pirogatetrahydrofuraniny, cyklohydrolazy 5-trifosforanu guanozyny itp.). Większość przypadków śmiertelnych przypadków odnotowano u pacjentów z rzadkimi zmianami w PKU, z klinicznymi objawami wszystkich postaci choroby podobnych. Ryzyko porodu ze zmutowanym genem hydroksylazy fenyloalaniny wzrasta, jeśli rodzice są bliskimi krewnymi (z blisko spokrewnionymi małżeństwami).
Diagnostyka
W przypadku podejrzenia choroby genetycznej diagnozę ustala się na podstawie agregatu danych uzyskanych w wyniku badania anamnezy choroby - danych genealogicznych, wyników klinicznych i medycznych badań genetycznych. Do szybkiego wykrycia chorób wrodzonych (PKU, mukowiscydoza, galaktozemia itp.) Program obowiązkowej masyBadanie laboratoryjne wszystkich noworodków (badanie przesiewowe noworodków).
Jeśli przyszli rodzice są świadomi obecności zmutowanego genu, nowoczesna medycyna oferuje sposoby wykrywania wady w czasie ciąży (prenatalna diagnoza płodu metodą inwazyjną). W celu podziału fenyloketonurii według rodzaju ciężkości stosuje się klasyfikację warunkową opartą na poziomie fenyloalaniny w płynie fibrynowym pochodzącym z plazmy plazmowej:
- Ciężka fenyloketonuria - 1200 mkmol /l.
- Średnia - 60-1200 μmol /l.
- Łatwy (nie wymaga obróbki) - 480 μmol /l.
Badanie przesiewowe
Wykrywanie nieprawidłowości genetycznych występuje w kilku etapach. W pierwszym etapie hospitalizacji u wszystkich dzieci w 3-5 dni życia pobierane są próbki krwi obwodowej (na pięć) do badań. Materiał jest nakładany na papierową formę i przesyłany do laboratorium biochemicznego, gdzie odbywa się jego analiza biochemiczna. W drugim etapie badania przesiewowego określa się stężenie fenyloalaniny o wartości normalnej.

Jeżeli nie wykryto żadnych zmian patologicznych, diagnoza kończy się, co zapisano na karcie dziecka. W przypadku odchyleń od normy, wyniki diagnozy są wysyłane do lekarza-pediatry, aby zapewnić udoskonalone badanie próbki krwi noworodka. Zdrowie dziecka zależy od terminowego i dokładnego wykonania wszystkich działań mających na celu wykrycie nieprawidłowości. Jeśli diagnoza zostanie potwierdzona po powtórnym teście przesiewowym, rodzice dzieckawysłany do kliniki na potrzeby genetyki dziecięcej w celu leczenia.
Analizy i badania potwierdzające diagnozę
Powtórna diagnoza w przypadku wykrycia odchylenia od normy podczas wstępnego testu przesiewowego jest przeprowadzana poprzez ponowną ocenę analiz. Oprócz oznaczania zawartości fenyloalaniny we krwi w metodach diagnostycznych PKU u dzieci i dorosłych obejmują:
- Próba ścinająca - oznaczanie kwasu fenylopirogronowego w moczu przez dodanie chlorku żelaza do biomateriału (występuje niebiesko-zielone zabarwienie);
- Test Gatree - ocena stopnia reakcji mikroorganizmów na produkty metaboliczne lub enzymy zawarte w krwi pacjenta;
- chromatografia - badanie właściwości chemicznych substancji rozproszonych między dwiema fazami;
- fluorymetria - naświetlanie biomateriału za pomocą promieniowania monochromatycznego w celu określenia stężenia zawartych w nim substancji;
- elektroencefalografia - diagnoza elektrycznej aktywności mózgu;
- obrazowanie rezonansu magnetycznego - naruszenie jądra atomowego komórek za pomocą fal elektromagnetycznych i pomiar ich odpowiedzi.
Leczenie klasycznego fenyloketonurium
Podstawą terapii fenyloketonurią jest ograniczenie spożycia produktów, które są źródłem białek pochodzenia zwierzęcego i roślinnego. Jedynym sposobem skutecznego leczenia jest terapia dietetyczna, której adekwatność szacowana jest na podstawie zawartości fenyloalaniny w surowicy. Maksymalny dopuszczalny poziom aminokwasów u pacjentów w różnych grupach wiekowychjest:
- u noworodków i dzieci poniżej 3 lat - do 242 μmol /l;
- dla przedszkolaków - do 360 mkmol /l;
- u pacjentów w wieku od 7 do 14 lat - do 480 μmol /L;
- u młodzieży - do 600 μmol /l.
Wpływ diety zależy od tego, na którym etapie choroby następuje korekta diety. We wczesnej diagnozie wrodzonej patologii, dieta jest zalecana od 8 tygodnia życia (po tym okresie już zaczynają się nieodwracalne zmiany). Brak szybkich pomiarów prowadzi do powikłań i obniżenia poziomu inteligencji o 4 punkty w ciągu 1 miesiąca od narodzin do początku leczenia.

Biorąc pod uwagę, że dieta terapeutyczna dla fenyloketonurii obejmuje całkowite wykluczenie z diety białka zwierzęcego, konieczne jest stosowanie innych źródeł niezbędnych aminokwasów, a także witamin z grupy B, związków mineralnych zawierających wapń i fosfor. Produkty przeznaczone do dodania do diety niebiałkowej obejmują:
- hydrolizaty białkowe (Amygens, Aminazole, fibrynozy);
- nie zawierają mieszanin fenyloalaniny nasyconych z niezbędnymi aminokwasami - Tetrafen, bez fenylu.
Oprócz środków leczniczych mających na celu wyeliminowanie przyczyny funkcjonowania organizmu należy zastosować leczenie objawowe mające na celu wyeliminowanie wad mowy i normalizację koordynacji ruchów. Kompleksowa terapia obejmuje zabiegi fizjoterapeutyczne, masaż, pomoc logopedy, psychologa, wykonywanie ćwiczeń gimnastycznych. W niektórych przypadkach przy terapii dietetycznejpokazuje stosowanie leków przeciwdrgawkowych, nootropowych i naczyniowych.
Cechy leczenia nietypowych postaci
fenyloketonuria II i typu III nieuleczalne nyzkobelkovoy diety - poziom fenyloalaniny we krwi pozostaje na stałym poziomie, przy jednoczesnym ograniczeniu przepływu białka w organizmie lub objawów klinicznych przebiega nawet przy niższych aminokwasów. Skuteczną terapię tych postaci choroby prowadzi się przy pomocy:
- tetrahydrobiopteryna - czynnik dotkniętego enzymem;
- syntetyczne analogi tetrahydrobiopteryny - te substancje są lepiej penetrowane przez barierę krew-mózg;
- leki terapia zastępcza - nie usuwa fenyloketonurię przyczyny, ale utrzymać normalne funkcjonowanie organizmu (dawkowania lewodopy razem z Karbydofoy, oksytryptofan 5, 5 formyltetrahydrofolat);
- hepatoprotekcje - wspomagają funkcjonowanie wątroby;
- leki przeciwdrgawkowe;
- wprowadzenie genu hydroksylazy fenyloalaniny w wątrobie - metoda eksperymentalna.
Funkcje odżywiania noworodka i dieta
w pierwszym roku życia z PKU dopuszczalnych breastfeeds, ale ilość powinna być ograniczona. W wieku 6 miesięcy, z akceptowalnym poziomem wlotu fenyloalaniny 60-90 mg na 1 kg masy ciała dziecka (100 g mleka zawiera 5,6 mg fenyloalaninę). Począwszy od 3 miesięcy, dieta dziecka powinna być stopniowo powiększana poprzez wprowadzanie soków owocowych i tłuczonych ziemniaków.
Dzieci w wieku od 6 miesięcy wpuszczona do diety tłuczone warzyw, zbóż (z sago) bezbilkovyhpocałunki Po 7 miesiącach dziecku można podawać nyzkobilkovi makaron, 8 miesięcy - Chleb, który zawiera białko. Nie ustalono wieku, w którym należy ograniczyć przenoszenie białka w ciele chorego dziecka. Lekarze prowadzone dotąd dyskusje na temat celowości kształcenia diecie, ale zgadzają się, że co najmniej 18 lat, aby stosować się do diety.
fenyloketonuria zdiagnozowano u kobiet nie jest powodem do rezygnacji z urodzeniem dziecka. Kobiety w ciąży z PKU, aby uniknąć uszkodzenia płodu podczas ciąży i zapobiegania możliwych komplikacji jest konieczne przed planowanym koncepcji i podczas ciąży diety niemowląt z ograniczeniem fenyloalaniny (jego poziom we krwi powinien wynosić do 242 mmol /l).
Mieszanki szczepionki dla niemowląt
Dieta dla fenyloketonurię podstawie znacznego zmniejszenia dawki naturalnego błonnika w codziennej diecie, ale ciało noworodka może rozwijać się normalnie w przypadku braku niezbędnych mikroelementów. W celu wypełnienia tych potrzeb laktozy niemowląt stosować w mieszaninie aminokwasowej białek, które, zgodnie z prawem Rosyjskiej, pacjenci muszą być wolne od opłat.
Tolerancja niemowlęcia na fenyloalaninę w pierwszym roku życia szybko się zmienia, dlatego konieczne jest kontrolowanie jego stężenia we krwi dziecka i dostosowywanie diety. Miksy są dla określonych grup wiekowych:
- dla niemowląt do roku wyznaczonego na Affenilak 15, Analog-JV, PKU-1, PKU-mix, PKU Anamix;
- dla dzieci w wieku powyżej 1 roku życiarok przepisać wzbogacony w witaminy i minerały mieszankę o wysokiej zawartości białka - PKU Prima, P-AM Universal, PKU-1, PKU-2, HRM Maxamed, HR Maxumum.

Produkty dietetyczne do uzupełniania zapasów białka
Jednym z głównych składników diety dietetycznej dla fenyloketonurii jest żywność niskobiałkowa na bazie skrobi. Suplementy te zawierają hydrolizat kazeiny, tryptofanu, tyrozyny, metioniny, azotu i zapewniają dziecku codzienne zapotrzebowanie na białko dziecka, które jest niezbędne dla prawidłowego rozwoju i wzrostu. Specjalistyczne produkty, uzupełniające brak niezbędnych minerałów i aminokwasów w braku ich diety, to:
- Berlofen;
- Tsimorgan;
- Minafen;
- Apotoni.
Dieta dla dzieci w wieku przedszkolnym i dzieci w wieku szkolnym
Ponieważ organizm dostosowuje się do fenyloalaniny u dzieci w wieku od 5 lat, możliwe jest stopniowe ograniczenie ograniczeń dietetycznych. Rozszerzenie diety polega na wprowadzeniu zbóż, produktów mlecznych, produktów mięsnych. Uczniowie szkół średnich mają już wysoką tolerancję na fenyloalaninę, więc w tym wieku można kontynuować rozszerzanie diety, podczas gdy konieczne jest monitorowanie reakcji na wszystkie zmiany w diecie. Do monitorowania stanu dziecka stosuje się następujące metody:
- ocena parametrów neurologicznych, stanu psychicznego;
- kontrola parametrów elektroencefalogramu;
- określenie poziomu fenyloalaniny.
Grupy produktów w PKU
W diecie pacjentów z PKU wraz z białkami niebędącymi białkamiProdukty skrobiowe i mieszaniny terapeutyczne obejmują również produkty pochodzenia naturalnego. Podczas sporządzania menu należy wyraźnie obliczyć ilość spożywanego białka i nie przekraczać zalecanego lekarza dozującego. Aby wykluczyć skutki toksyczne na ciele, opracowywane są 3 listy produktów, które zawierają niedozwolone (czerwone), niereprezentowane (pomarańczowe) i dozwolone (zielone) pozycje.
Czerwona lista
Fenyloketonuria rozwija się na tle braku enzymu, który przekształca się w fenyloalaninę tyrozyny, więc wysoka zawartość białka jest podstawą do przypisania produktów do listy zakazanej (czerwonej). Pozycje z tej listy powinny całkowicie wykluczać dietę pacjenta na PKU:
- mięso;
- narządy wewnętrzne zwierząt, produkty uboczne;
- kiełbasy, kiełbasy;
- owoce morza (w tym ryby);
- jaj wszystkich ptaków;
- produkty z kwaśnego mleka;
- orzechy;
- rośliny strączkowe i zboża;
- produkty sojowe;
- galaretowate naczynia;
- słodycze;
- aspartam.
Pomarańczowa lista
Produkty wprowadzane do ciała dziecka, u którego zdiagnozowano PKU, znajdują się na pomarańczowej liście. Włączenie do diety przedmiotów z tej listy jest dopuszczalne, ale w ściśle ograniczonych ilościach. Chociaż te produkty nie zawierają dużo białka, ale mogę również zwiększyć poziom fenyloalaniny, dlatego ich stosowanie nie jest zalecane:
- warzywa w puszkach;
- dania ziemniaczane i ryżowe;
- kapusta;
- mleko;
- sorbet.
Zielona lista
Produkty niebiałkowe są dopuszczone do stosowania u pacjentów z fenyloketonurią bez żadnych ograniczeń. Przed zakupem przedmiotów z zielonej listy należy zbadać skład wskazany na opakowaniu i upewnić się, że nie ma barwnika aspartamu zawierającego fenyloalaninę:
- owoce;
- warzywa (z wyjątkiem ziemniaków i kapusty);
- jagody;
- zieleni;
- zboża skrobiowe (sago);
- miód, cukier, dżem;
- produkty mączne z mąki kukurydzianej lub ryżowej;
- masło, tłuszcze (masło, słonecznik, oliwka).
Jak kontrolować poziom fenyloalaniny we krwi
Fenyloketonuria jest chorobą nieuleczalną, która może zostać przekształcona w fazę stagnacji poprzez zastosowanie terapii dietetycznej oraz środków terapeutycznych i profilaktycznych. Kiedy zmiana warunków życia, naruszenie diety, choroba może się ponownie zaostrzyć, więc pacjenci potrzebują obserwacji przez całe życie. Proces kontrolny polega na okresowym określaniu poziomu fenyloalaniny we krwi. Częstotliwość dostaw zależy od wieku pacjenta:
- do 3 miesięcy - badanie krwi należy przeprowadzać co tydzień, aż do uzyskania stabilnych wyników;
- od 3 miesięcy do 1 roku - 1-2 razy w miesiącu;
- od 1 do 3 lat - 1 raz w ciągu 2 miesięcy;
- w ciągu 3 lat - raz na kwartał.

Krew do analiz wydaje się być 3-4 godziny po jedzeniu. Oprócz badań przesiewowych rozwój PKU jest kontrolowany poprzez określenie stanu odżywienia, fizycznego, emocjonalnego rozwoju pacjenta, poziomu zdolności intelektualnychi rozwój języka. Zgodnie z wynikami obserwacji może być konieczna dodatkowa diagnostyka z udziałem odpowiednich specjalistów.
Filmy wideo
Informacje zawarte w tym artykule mają charakter informacyjny. Materiały artykułu nie wymagają niezależnego leczenia. Tylko wykwalifikowany lekarz może zdiagnozować i udzielić porady dotyczącej leczenia na podstawie indywidualnych cech konkretnego pacjenta.














